1. Datasets and preliminaries
GLIMPSE is a tool of software for imputation and phasing of low-coverage datasets in the form of genotype gikelihoods (GLs) at all variant positions.
GLIMPSE requires HTSlib and BCFtools v1.7 (or later) as a requirement, since it makes use of indexed VCF/BCF files.
Here we also used BCFtools to compute genotype likelihoods.
In this tutorial, we show how to run GLIMPSE from sequencing reads data (BAM/CRAM file) to obtain refined genotype and haplotype calls.
The data and the scripts for this tutorial can be downloaded HERE.
A minimal set of data to run this pipeline can be found in the folder tutorial_hg19. Here we assume that the folder is located as a subfolder of the GLIMPSE installation: tutorial_chr19.
All datasets used here are in GRCh37/hg19 genome assembly
A dataset containing chromosome 22 downsampled 1x sequencing reads (BAM file) for 1 individual (NA12878) from ASW population is provided with this tutorial,
together with a reference genome FASTA file needed for genotype likelihood computations.
1.1. Setting the environment and binaries
All scripts have been written assuming > cd GLIMPSE/tutorial_hg19/ as current directory. Therefore, we require to move to the tutorial_hg19 directory:
> cd GLIMPSE/tutorial_hg19/
From this folder we build all the GLIMPSE software needed (chunk, phase, ligate, sample and concordance). For this reason, we require that GLIMPSE can correctly compile on your machine and you might be required to edit the Makefiles manually. See installation instructions if there are any problems at this stage. We created a script to configure a directory containing symbolic links to the GLIMPSE binaries we will need later. To run the setup script, simply run:
> ./step1_script_setup.sh
If the script runs correctly, the software compiles and the bin folder containing symbolic links to the binaries has been created, everything is correctly setup.
1.2. Low coverage reads
In this example we downsampled the publicly available 30x data available for NA12878 provided by the Genome In A Bottle consortium (GIAB). We downloaded the full dataset, kept only chromosome 22 reads and downsampled to 1x. The resulting BAM file can be found in:
GLIMPSE/tutorial_hg19/NA12878_1x_bam/NA12878.chr22.1x.bam[.bai]
Details of how we downsampled the dataset are provided in appendix section A1.1.
2. Reference panel preparation
In order to run accurate imputation we recommend to use a targeted reference with ancestry related to the target samples, if possible. For this tutorial we use the 1000 Genomes Project phase 3 reference panel. Since the reference panel contains data of our target sample, we need to remove it from the reference panel. This steps downloads the 1000 genomes GRCh37/hg19 data from the EBI ftp site.
2.1. Download of the reference panel files
The 1000 Genomes phase 3 reference panel is publicly available at EBI 1000 genomes ftp site. We can download chromosome 22 data using:
> wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz{,.tbi}
2.2. Remove NA12878 from the reference panel and perform basic QC
We used BCFtools to remove sample NA12878 from the reference panel and we export the dataset in BCF file format (for efficiency reasons). We also performs a basic QC step that includes keeping only SNPs and remove multiallelic records. The original reference panel files are then deleted from the main tutorial_hg19 folder:
> CHR=22
> bcftools view -m 2 -M 2 -v snps -s ^NA12878 ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz -Ob -o reference_panel/1000GP.chr22.noNA12878.bcf
> reference_panel/1000GP.chr22.noNA12878.bcf
> bcftools index -f reference_panel/1000GP.chr22.noNA12878.bcf
> rm ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz*
The resulting reference panel files can be found in:
GLIMPSE/tutorial_hg19/reference_panel/1000GP.chr22.noNA12878.bcf[.csi]
The reference panel can now be used for genotype likelihoods calculations and imputation.
3. Computation of genotype likelihoods
GLIMPSE requires input data to take the form of Genotype Likelihoods (GLs). GLs need to be computed at all target individuals and all variant sites present in the reference panel of haplotypes used for the imputation. In this section, we describe a simple procedure to compute GLs from sequencing data using BCFtools. However, other callers, such as GATK for instance, can be also considered as soon as the GLs are encoded in a VCF/BCF file using the FORMAT/PL field.
Please note that BCFtools does not call correctly genotype likelihoods for indels and this might affect the quality of the imputation even at non-indels. For this reason in this tutorial we only focus on bi-allelic SNPs.
For this tutorial we only follow step 2.1 and step 2.2, because step 2.3 is not necessary since we have only one target sample in our study panel.
The code for sections 2.1 and 2.2 is included in the script step2_script_GL.sh.
3.1. Extracting variable positions in the reference panel
We want this information to tell BCFtools at which positions to make a call.
Since BCFtools does not seem to compute correctly genotype likelihood for indels, here we only focus on SNPs (however, GLIMPSE can impute any type of variants as soon it is bi-allelic and has GLs being properly defined).
To perform the extraction from the chromosome 22 of a reference panel 1000GP.chr22.noNA12878.bcf, run first bcftools as follows:
> bcftools view -G -m 2 -M 2 -v snps reference_panel/1000GP.chr22.noNA12878.bcf -Oz -o reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz
> bcftools index -f reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz
Then, convert the output file 1000GP.chr22.noNA12878.sites.vcf.gz into TSV format and index the file using tabix (requires htslib in PATH), using this command:
> bcftools query -f'%CHROM\t%POS\t%REF,%ALT\n' reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz | bgzip -c > reference_panel/1000GP.chr22.noNA12878.sites.tsv.gz
> tabix -s1 -b2 -e2 reference_panel/1000GP.chr22.noNA12878.sites.tsv.gz
In a general pipeline, we should generate this pair of files (*.vcf.gz + *.tsv.gz) for all chromosome separately.
3.2. Computing GLs for a single individual at specific positions
Once ready the position files, we can then start doing the calling in itself.
We use the downsampled 1x BAM file generated in section 1.1 NA12878_1x_bam/NA12878.chr22.1x.bam.
BCFtools requires the reference genome in order to align the BAM file. We used the reference genome version used by the 1000 Genomes Project (reference_genome/human_g1k_v37.fasta.gz[fai])
We can run the following command:
> BAM=NA12878_1x_bam/NA12878.chr22.1x.bam
> VCF=reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz
> TSV=reference_panel/1000GP.chr22.noNA12878.sites.tsv.gz
> REFGEN=reference-genome/human_g1k_v37.chr22.fasta.gz
> OUT=NA12878_1x_vcf/NA12878.chr22.1x.vcf.gz
> bcftools mpileup -f ${REFGEN} -I -E -a 'FORMAT/DP' -T ${VCF} -r 22 ${BAM} -Ou | bcftools call -Aim -C alleles -T ${TSV} -Oz -o ${OUT}
> bcftools index -f ${OUT}
Note here, that we use -T reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz in the first part of the command line and -T reference_panel/1000GP.chr22.noNA12878.sites.tsv.gz in the second part of the command line.
You may also tune the options of BCFtools to your specific needs, requirements and data.
As an output of this step we have a VCF file format containing genotype likelihoods at each variable position in the reference panel.
3.3. Merging genotype likelihoods of multiple individuals
In the case of multiple target individuals, an additional step, not required in this pipeline, is to generate a single file containing genotype likelihoods for all target individuals. This can be easily done using BCFtools, for example:
> bcftools merge -m none -r 22 -Oz -o merged.chr22.1x.vcf.gz -l list.txt
Where list.txt is a text file containing the full list of VCF/BCF files containing GLs of each target individual in the study, one individual file per line.
The resulting file must be indexed and can be used in the subsequent steps of the pipeline.
4. Split the genome into chunks
One important step prior is to define the chunks where to run imputation and phasing. This step is not trivial because different long regions increase the running time, but small regions can drastically decrease accuracy. Also there are several variables to take into account: the amount of missingness in the defined region and the length in Mb. For these reasons we developed a tool in the GLIMPSE suite (GLIMPSE_chunk) that is able to quickly generate imputation chunks taking into account all this information.
4.1. Chunking a chromosome
We use GLIMPSE_chunk to generate imputation regions for the full chromosome 22, modifying few default parameters in this way:
> bin/GLIMPSE_chunk --input reference_panel/1000GP.chr22.noNA12878.sites.vcf.gz --region 22 --window-size 2000000 --buffer-size 200000 --output chunks.chr22.txt
5. Impute and phase a whole chromosome
The core of GLIMPSE is the GLIMPSE_phase method. The algorithm works by iteratively refining the genotype likelihoods of the target individuals in the study. The output of the method is a VCF/BCF file containing:
- the best guess genotype in the FORMAT/GT field
- the imputed genotype dosage in the FORMAT/DS field
- the genotype probabilities in the FORMAT/GP field
- the last (max 15) sampled haplotypes in the FORMAT/HS field
5.1. Running GLIMPSE
To run GLIMPSE_phase we only need to run a job for each imputation chunk. Each job runs on 1 thread in this example. Input data are the dataset containing the genotype likelihoods, a reference panel of haplotypes, a fine-scale genetic map (e.g. from the International HapMap project) and buffered and imputation regions. We run GLIMPSE phase using default parameters that can apply to most datasets. For extremely low coverage and small reference panels, increasing the number of iterations might give more accuracy.
> VCF=NA12878_1x_vcf/NA12878.chr22.1x.vcf.gz
> REF=reference_panel/1000GP.chr22.noNA12878.bcf
> MAP=../maps/genetic_maps.b37/chr22.b37.gmap.gz
> while IFS="" read -r LINE || [ -n "$LINE" ];
> do
> printf -v ID "%02d" $(echo $LINE | cut -d" " -f1)
> IRG=$(echo $LINE | cut -d" " -f3)
> ORG=$(echo $LINE | cut -d" " -f4)
> OUT=GLIMPSE_imputed/NA12878.chr22.1x.${ID}.bcf
> bin/GLIMPSE_phase --input ${VCF} --reference ${REF} --map ${MAP} --input-region ${IRG} --output-region ${ORG} --output ${OUT}
> bcftools index -f ${OUT}
> done < chunks.chr22.txt
6. Ligate multiple chunks together
Merging together different chunks of the sample chromsome is a standard procedure and it is usually straightforward in the case of genotype data. However GLIMPSE phase generates phased information in addition of genotype information. In this case, ligation is a required step to merge multiple chunks together without losing phased imputation. For this purpose we developed a software in the GLIMPSE software suite, called GLIMPSE_ligate. The output of this step is a chromosome wide VCF/BCF file containing information coherent phasing information, stored in the FORMAT/HS field
6.1. Ligate whole chromosome chunks
GLIMPSE_ligate only requires a list containing the imputed files that need to be merged together:
> LST=GLIMPSE_ligated/list.chr22.txt
> ls GLIMPSE_imputed/NA12878.chr22.imputed.*.bcf > ${LST}
> OUT=GLIMPSE_ligated/NA12878.chr22.merged.bcf
> bin/GLIMPSE_ligate --input ${LST} --output $OUT
> bcftools index -f ${OUT}
In the case phased information is not needed, this can be the final step, providing chromosome-wide genotype information in the FORMAT/DS and FORMAT/GP field.
7. Sample haplotypes
In the case haplotype-level information is required, GLIMPSE_sample can sample from the FORMAT/HS field accurate, consensus based haplotypes. The output of this step is a VCF/BCF file containing information at the chromosome level, where the GT field contains accurate phased information.
7.1. Sample whole chromosome data
There are two main modes for GLIMPSE_sample can operate.
The first mode can sample haplotypes based on the probabilities given from the FORMAT/HS field (--sample), while the second mode outputs the most likely haplotypes given the values in the FORMAT/HS field (--solve).
We run GLIMPSE_sample using the --solve mode:
> VCF=GLIMPSE_ligated/NA12878.chr22.merged.bcf
> OUT=GLIMPSE_phased/NA12878.chr22.phased.bcf
> bin/GLIMPSE_sample --input ${VCF} --solve --output ${OUT}
> bcftools index -f ${OUT}
From the output dataset, accurate phased information, stored in the GT field can be directly used.
8. Check imputation accuracy
Since we downsampled the reads from the original 30x data, we might be interested now in checking how accurate the imputation is, compared the original 30x dataset. For this purpose we use the GLIMPSE_concordance tool, which can be used to compute the r2 correlation between imputed dosages (in MAF bins) and highly-confident genotype calls from the high-coverage dataset.
8.1. Running GLIMPSE_concordance
The GLIMPSE_concordance requires tool requires a file (--input) indicating:
- the region of interest;
- a file containing allele frequencies at each site, in this case the 1000 Genomes reference panel (NOT the ideal choice as the AF are from different populations - other datasets such as GnomAD should be preferred);
- the validation dataset called at the same positions as the imputed file (in a similar way as showed in appendix A1, without the downsampling);
- the imputed data.
concordance.lst having all the correct files in the right order for this tutorial.
Other parameters specify how confident we want a site to be in the validation data and the MAF bins.The GLIMPSE_concordance too can be run as follows:
> ./bin/GLIMPSE_concordance --input concordance.lst --minDP 8 --output GLIMPSE_concordance/output --minPROB 0.9999 \
> --bins 0.00000 0.00100 0.00200 0.00500 0.01000 0.05000 0.10000 0.20000 0.50000ex \
> --thread 4
8.2. Visualising the results
The GLIMPSE_concordance folder contains several files describing the quality of the imputation. In particular, we are interested in the file GLIMPSE_concordance/output.rsquare.grp.txt.gz.
We can visualise the results by going in the plot folder and running:
> ./concordance_plot.py
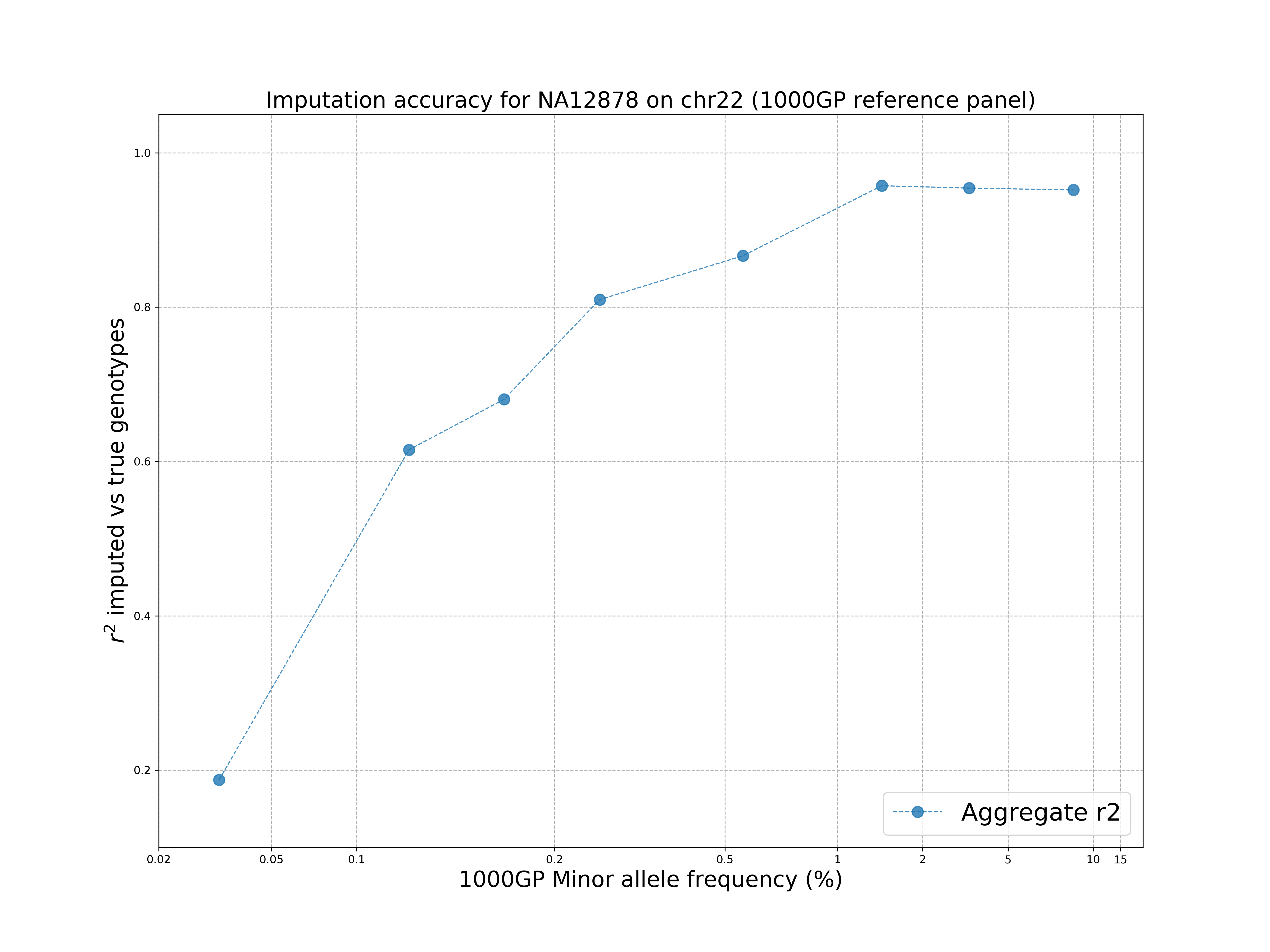
The command requires python3 and matplotlib installed.
The plot shows that the 1000 Genomes reference panel can be used to impute accurately variants up to ~1% MAF and there is a drop at rare variants, as expected. The plot does not appear very smooth due to the poor choice of the allele frequencies (1000 Genomes project).
A1. Low coverage reads
A1.1. Downsample GIAB high coverage data
We can donwload the dull downsampled (30x) dataset for NA12878 from NCBI website, provided by Genome In A Bottle consortium:
> wget ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/NA12878/NIST_NA12878_HG001_HiSeq_300x/RMNISTHS_30xdownsample.bam{,.bai}
The size of this dataset is in the order of 150GB, so it can be prohibitive on some systems. Once the BAM files have been downloaded, we only keep chromosome 22 and re-index the dataset:
> samtools view -bh RMNISTHS_30xdownsample.bam 22 -o NA12878.chr22.30x.bam
> samtools index NA12878.chr22.30x.bam
> rm RMNISTHS_30xdownsample.bam*
Finally, we can downsample the 30x data to low coverage (1x) using samtools
> samtools view -s $FRAC 0.030973196415286 -bo NA12878_1x_bam/NA12878.chr22.1x.bam NA12878.chr22.30x.bam
> samtools index NA12878_1x_bam/NA12878.chr22.1x.bam
> rm NA12878.chr22.30x.bam*
The output dataset NA12878_1x_bam/NA12878.chr22.1x.bam is the file provided with GLIMPSE.